Marker gene studies often use short amplicons spanning one or more hypervariable regions from an rRNA gene to interrogate the community structure of uncultured environmental samples. Although the target regions are chosen for their discriminatory power, the short length of high-throughput sequencing reads limits the phylogenetic signal of the data. This is particularly unfortunate in the study of microscopic eukaryotes where horizontal gene flow is limited and the rRNA gene is expected to accurately reflect the species phylogeny.
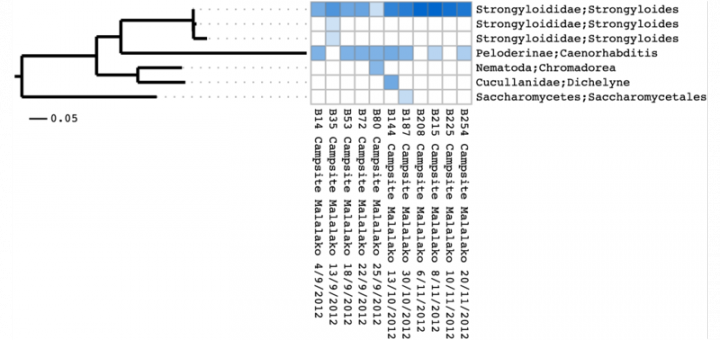
A promising alternative to de novo phylogenetic analysis of amplicon data is to build a reference phylogeny based on sequence data from the complete marker gene and iteratively extend the tree with the short sequences from the metagenetic samples under study. Based on this approach, we built Séance, a community analysis pipeline focused on the analysis of the 18S marker gene. S ́eance combines the alignment extension and phylogenetic placement capabilities of the Pagan multiple sequence alignment program with a suite of tools to preprocess, cluster and visualise datasets composed of many samples. In collaboration Jukka Jernvall’s group, we have applied Séance on the analysis of 454 data from a longitudinal study of intestinal parasite communities in wild rufous mouse lemurs (Microcebus rufus) from Ranomafana National Park in southeast Madagascar.