Active research projects
Template-switch mutations and evolution of RNA genes
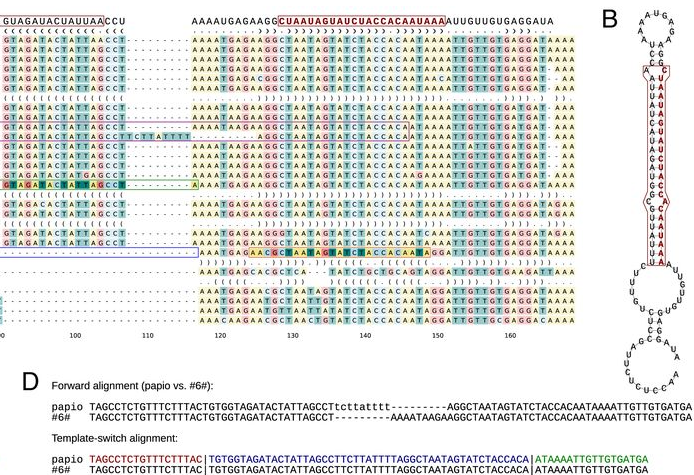 Template switching in DNA replication was proposed as the mechanism converting near-perfect palindromic repeats into perfect palindromic repeats in bacteria. We extended this model to allow for any kind of local switch events and implemented it in a computational tool. Applying the method to genomic alignments of human and chimp, we identified large numbers of mutation patterns consistent with the template-switch process, some of them of a previously unknown type (publication). Following that, we applied the method to modern high-throughput data and showed that each human carries thousands of template-switch loci. Strikingly, we observed that commonly-used analysis tools may hide the signals of these mutations such that they may go unnoticed e.g. in medical genetic studies (publication). We continue this project with the analyses of somatic mutation data.
Template switching in DNA replication was proposed as the mechanism converting near-perfect palindromic repeats into perfect palindromic repeats in bacteria. We extended this model to allow for any kind of local switch events and implemented it in a computational tool. Applying the method to genomic alignments of human and chimp, we identified large numbers of mutation patterns consistent with the template-switch process, some of them of a previously unknown type (publication). Following that, we applied the method to modern high-throughput data and showed that each human carries thousands of template-switch loci. Strikingly, we observed that commonly-used analysis tools may hide the signals of these mutations such that they may go unnoticed e.g. in medical genetic studies (publication). We continue this project with the analyses of somatic mutation data.
An intriguing property of template-switch mutations is their ability to create perfect reverse-complement repeats with one mutation event. These repeats can form hairpin structures which then are the basis of all RNA gene secondary structures. Studying the RNA gene evolution in primates and other mammals, we showed that the template-switch mechanism can explain the evolution of RNA secondary structures through compensatory mutations (publication). We then extended this analysis and showed the mechanism can also create entirely novel microRNA genes (publication).
We are currently developing analysis software for identification of template switch events in clinical genetics and medical research data. The project (2025-2026) is funded by Research Council of Finland.
Past research projects
Pinniped population genetics and speciation
![]() We are members of the Saimaa Ringed Seal Genome Project (project page) and involved in the analysis of the whole-genome sequencing data. We have studied the evolutionary history and population structure of the species (publication) and our aims is to understand the genomic effects of the isolation and its recent population bottleneck. We were involved in a study proposing the species status for the Saimaa seals (news release, publication), which they subsequently obtained (news release). We are generally interested in the evolution of pinnipeds and were involved in the identification of a genetically and phenotypically distinct seal population in the Arctic (publication). We are continuing on this topic with the aim of understanding the pinniped speciation processes and the genetic basis of their rapid phenotypic evolution.
We are members of the Saimaa Ringed Seal Genome Project (project page) and involved in the analysis of the whole-genome sequencing data. We have studied the evolutionary history and population structure of the species (publication) and our aims is to understand the genomic effects of the isolation and its recent population bottleneck. We were involved in a study proposing the species status for the Saimaa seals (news release, publication), which they subsequently obtained (news release). We are generally interested in the evolution of pinnipeds and were involved in the identification of a genetically and phenotypically distinct seal population in the Arctic (publication). We are continuing on this topic with the aim of understanding the pinniped speciation processes and the genetic basis of their rapid phenotypic evolution.
Stickleback population genetics and evolutionary mechanisms
![]() We have a long-time collaboration with prof Juha Merilä on genomic analyses of stickleback fishes. We are interested in understanding the evolution and the history of different pond and marine populations, especially the genomic effects of small founder populations and drift within ponds. More information about our stickleback research at the publications page.
We have a long-time collaboration with prof Juha Merilä on genomic analyses of stickleback fishes. We are interested in understanding the evolution and the history of different pond and marine populations, especially the genomic effects of small founder populations and drift within ponds. More information about our stickleback research at the publications page.
Advanced web application for evolutionary sequence analysis
 Viewing multiple sequence alignments with the associated sequence phylogeny helps to understand the alignment and draw conclusions from it. Wasabi is a graphical interface for evolutionary sequence analysis and data visualisation. It is built upon HTML5 and Javascript technologies and so can be run inside any modern web browser. Wasabi can be run locally as a graphical interface to underlying analysis software or launched from a remote server to offload the computational heavy-lifting. You can start using Wasabi here.
Viewing multiple sequence alignments with the associated sequence phylogeny helps to understand the alignment and draw conclusions from it. Wasabi is a graphical interface for evolutionary sequence analysis and data visualisation. It is built upon HTML5 and Javascript technologies and so can be run inside any modern web browser. Wasabi can be run locally as a graphical interface to underlying analysis software or launched from a remote server to offload the computational heavy-lifting. You can start using Wasabi here.
Phylogeny-aware sequence alignment and extension of existing sequence alignments
The phylogeny-aware alignment method PRANK has been found to be an excellent sequence aligner for comparative evolutionary analyses. It should be used with caution for phylogenetic analyses, however, as the underlying algorithm uses the phylogenetic information from a guide tree during the alignment procedure and the resulting multiple alignment will reflect this phylogenetic structure. We studied approaches to get around this issue and reduce the potential bias in phylogenetic analyses. More at the Canopy homepage.
Accurate alignment of large numbers of sequences is demanding and the computational burden is further increased by downstream analyses depending on these alignments. We developed a phylogeny-aware approach to add new sequences to existing alignments without their full re-computation and maintaining the relative matching of existing sequences. The same ideas can be used to extend reference alignments with fragmented sequences that contain relatively little information, e.g. in next-generation metagenomics analyses. The functionalities have been implemented in the PAGAN software and the Glutton and Séance analysis pipelines build around that.
Reference-based scaffolding of RNA-seq data
Transcriptomics data produced with high-throughput sequencing methods targets nearly exclusively gene regions of the genomes and can potentially provide an inexpensive approach for evolutionary and comparative analyses. The challenge is to assemble the reads to longer contigs and to identify the correct homologous sequences. We developed Glutton, a targeted approach that uses information from distantly related species to accurately reconstruct transcriptome for non-model organisms lacking a reference genome.
Metagenomic analysis of noisy sequence data
High-throughput sequencing platforms produce data with characteristic errors. We have developed an analysis software that can correct for these errors and allows for using the noisy data for evolutionary analyses. More at the Séance homepage.